Introduction to RNA Sequencing Bioinformatics
Setup
Approximate time: 20 minutes
Goals
- Connect to the HPC cluster via On Demand Interface
- Download data
Log into the HPC cluster’s On Demand interface
- Open a Chrome browser visit ondemand.cluster.tufts.edu
- Log in with your Tufts Credentials
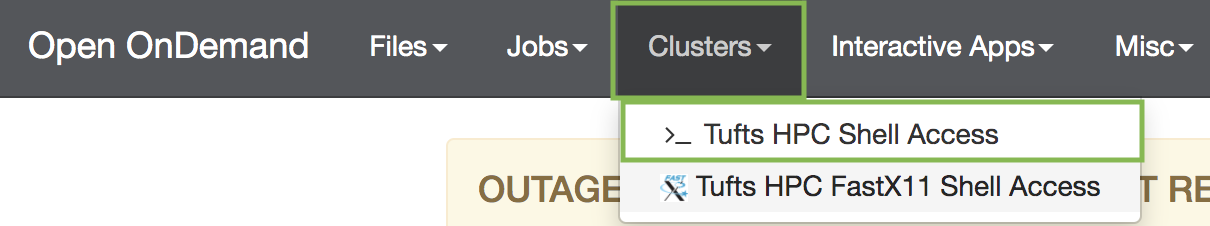
- On the top menu bar choose Clusters->HPC Shell Access
- Type your password at the prompt (the password will be hidden for security purposes):
whuo01@login.cluster.tufts.edu's password:
- You’ll see a welcome message and a bash prompt, for example for user
whuo01:
[whuo01@login001 ~]$
This indicates you are logged in to the login node.
- Type
clearto clear the screen
Compute node allocation
- Get an interactive session on a compute node by typing:
srun --pty -t 3:00:00 --mem 16G -N 1 -n 4 bash
Once you hit enter, you will see something like below showing that the job is queued:
[whuo01@login001 ~]$ srun --pty -t 3:00:00 --mem 16G -N 1 -n 4 bash
srun: job 55918493 queued and waiting for resources
If wait times are very long, you can try a different partitions by adding, e.g. -p interactive before bash.
Or, if you are you registered for the workshop, you can use following option before bash: -p preempt --reservation=bioworkshop.
This reservation will be available for one week after the workshop start.
You can press Ctrl-C to cancel your request and try again with different options, e.g.:
[whuo01@login001 ~]$ srun --pty -t 3:00:00 --mem 16G -N 1 -n 4 -p interactive bash
[whuo01@pcomp45 ~]$
The success is indicated by the change of node name after your username. Here it was changed from login001 to pcomp45.
This is an indication that you may proceed to the next step.
Note: If you go through this workshop in multiple steps, you will have to rerun this step each time you log in.
Course data
- Since our home directory will likely not have enough space for the analysis (> 3Gb), we’ll work in a course directory. Your work will be saved here for 30 days.** Change to the course directory
cd /cluster/tufts/bio/tools/training/intro-to-rnaseq/users/
**Note: If you have a project directory for your lab, you may use this instead.
These are located in /cluster/tufts with names like /cluster/tufts/labname/username/.
If you don’t know whether you have project space, please email tts-research@tufts.edu.
- Make a directory for your work (replace
whuo01in the below commands with your username)mkdir whuo01 cd whuo01 - Copy the course files into your own directory:
cp /cluster/tufts/bio/tools/training/intro-to-rnaseq/intro-to-RNA-seq-May-2020.tar.gz . - Unzip the course directory:
tar -xvzf intro-to-RNA-seq-May-2020.tar.gz - Take a look at the contents of the unzipped directory by typing:
tree intro-to-RNA-seq
Result:
intro-to-RNA-seq/
├── ERP004763_info.txt <-- sample description
├── raw_data <-- Folder with fastq files
│ ├── sample_info.txt
│ ├── SNF2
│ │ ├── ERR458500.fastq.gz <-- gzip compressed fastq files
│ │ ├── ERR458501.fastq.gz
│ │ ├── ERR458502.fastq.gz
│ │ ├── ERR458503.fastq.gz
│ │ ├── ERR458504.fastq.gz
│ │ ├── ERR458505.fastq.gz
│ │ └── ERR458506.fastq.gz
│ └── WT
│ ├── ERR458493.fastq.gz
│ ├── ERR458494.fastq.gz
│ ├── ERR458495.fastq.gz
│ ├── ERR458496.fastq.gz
│ ├── ERR458497.fastq.gz
│ ├── ERR458498.fastq.gz
│ └── ERR458499.fastq.gz
└── scripts <-- Folder with all commands
├── fastqc.sh
├── featurecounts.sh
├── intro.R
├── sbatch_star_align_individual.sh
├── sbatch_star_align.sh
└── sbatch_star_align_SNF2.sh
4 directories, 22 files
Data for the class
Publication: Statistical Models for RNA-seq Data Derived From a Two-Condition 48-replicate Experiment.
Purpose: The experiment seeks to compare a wild type Saccharomyces cerevisiae with a mutant that contains a knock-out in the gene SNF2. The purpose of the study is to analyze variability in sequencing replicates.
Project access number: PRJEB5348
Samples: The WT folder contains 7 sequencing files from a wild type yeast sample, SNF2 contains 7 sequencing files from a yeast sample with a knock-out mutation in the gene SNF2.
Note that for the workshop purposes we are treating the 7 sequencing files as if they originate from separate biological replicates.
Organism: Saccharomyces cerevisiae
Sequencing: Illumina HiSeq, Single End, 50bp read length
Workshop Schedule
- Course Home
- Introduction
- Currently at: Setup using Tufts HPC
- Next: Quality Control
- Read Alignment
- Gene Quantification
- Differential Expression
- Pathway Enrichment